When to Use nf-core/sarek and When Another Pipeline Might Be Better
August 11, 2025
·Fatima Farhan & Layla Bitar (Co-author)

Choosing the Right Bioinformatics Pipeline: Is nf-core/sarek the One for You?
Selecting the right bioinformatics pipeline is rarely a one-click decision. The same dataset can be processed in very different ways depending on your end goal: meeting strict regulatory requirements, preparing a manuscript for publication, or exploring patterns in an early-stage research project. The choice goes beyond simply looking at the type or size of your data. Factors such as the computational resources available, the technical expertise of your team, the need for reproducibility, institutional regulations, and even collaborative requirements all play a role. A pipeline that works perfectly in a well-funded research consortium might be impractical in a small clinical lab, and vice versa. This is why understanding not just what the pipeline can do, but how it fits into your specific environment, is critical before deciding.
One of the most common questions bioinformaticians face is: When should I use nf-core/sarek, and when might another pipeline be a better fit?
The answer depends on more than the dataset itself. Whether you are working in clinical care or in research exploration, your context will often determine whether nf-core/sarek is a perfect match or if you should consider alternatives.
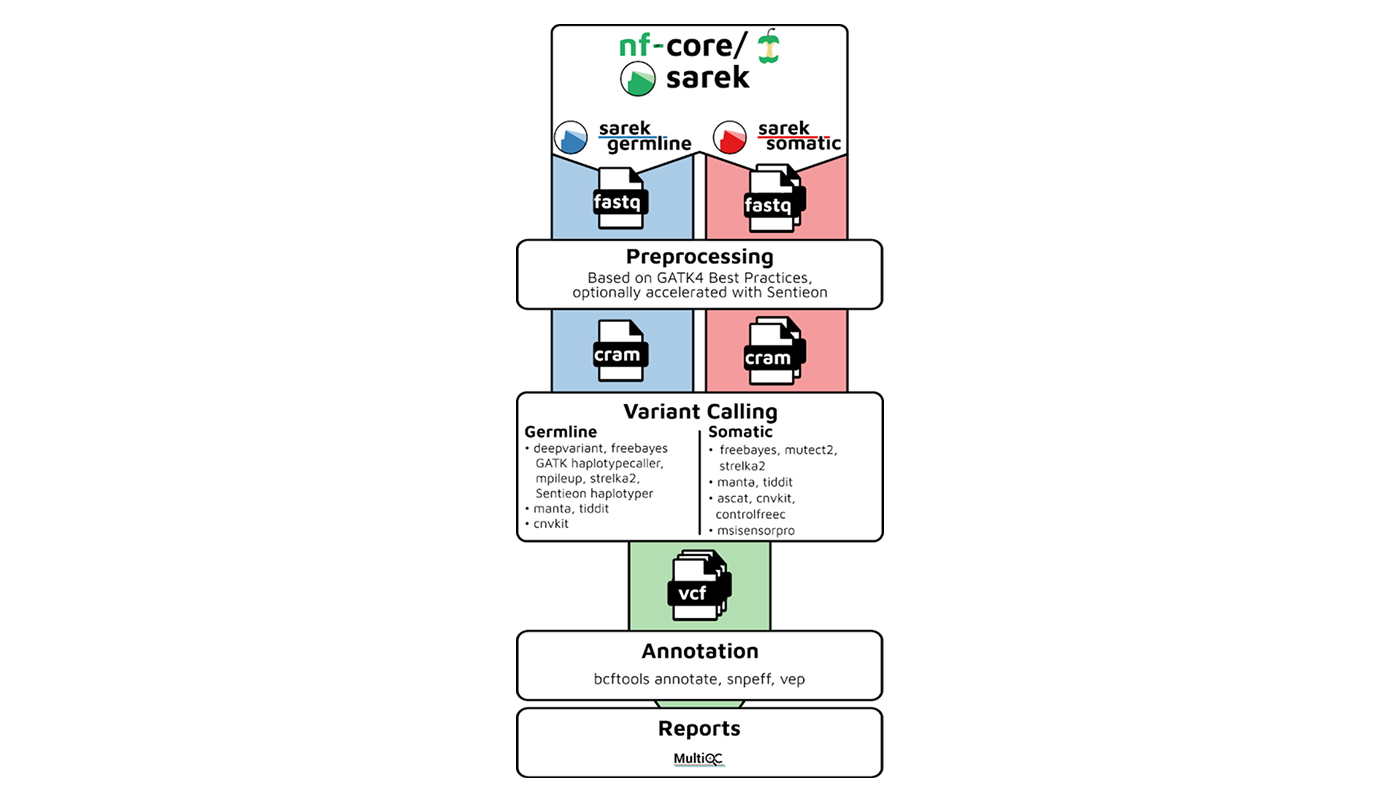
What Makes nf-core/sarek Stand Out
nf-core/sarek is part of the wider nf-core ecosystem: a collection of peer-reviewed, community-maintained pipelines built on Nextflow. Sarek is designed for germline and somatic variant calling on whole genome or whole exome data. It handles everything from alignment and quality control to variant annotation and report generation in a reproducible, multi-step workflow.
What sets it apart is its flexibility. Sarek supports multiple variant callers: Strelka2, Mutect2, HaplotypeCaller, and more, allowing you to tailor runs to your needs. Its modular design means you can fine-tune configurations, while built-in MultiQC integration gives you immediate insight into quality metrics. And because it is actively maintained, you benefit from regular updates, clear versioning, and a vibrant user community.
Research and Clinical Use Cases
The value of nf-core/sarek varies depending on whether it is used in a research or clinical setting. In research, reproducibility and flexibility often outweigh speed, while clinical workflows prioritize compliance, validation, and turnaround time. These differing priorities determine how the pipeline is configured, its suitability for a given project, and whether an alternative solution might be more effective.
Strengths and Limits in Research
For research labs, nf-core/sarek is often a first choice when reproducibility and collaboration matter. Because it is open source and standardized, different teams can run the same workflow and be confident in producing consistent results, critical for joint projects and publications.
Example: A cancer genomics lab studying tumor-normal pairs across 100 patients needs a robust variant-calling pipeline with reliable logging and multi-caller support. With nf-core/sarek, they can deploy a single, tested configuration for every run, ensuring consistency and transparency from start to finish.
However, it is not always the right fit. If you are working with ultra-low coverage datasets, highly specialized targeted panels, or tools that are not yet integrated into Sarek, you might be better served by alternatives such as Snakemake or bcbio-nextgen.
Strengths and Limits in Clinical Workflows
In clinical environments, Sarek’s strengths are its traceability, adaptability, and transparency. You can configure it to meet regulatory standards, provided your team performs its own validation. It works well for high-throughput WGS or WES in diagnostic workflows, especially where flexibility is needed.
Example: A diagnostics team screening for hereditary cancer variants uses a validated configuration of nf-core/sarek to comply with internal regulatory processes. They integrate it with downstream interpretation tools, enabling efficient, auditable variant reporting.
Still, some clinical labs opt for commercial tools such as DRAGEN or Sentieon when vendor support, out-of-the-box validation, or rapid turnaround is a priority. And if your lab lacks the resources to perform its own regulatory validation, an open-source pipeline may not be the best immediate choice.
Performance Considerations with nf-core/sarek
Beyond features and compatibility, performance is often the deciding factor for many teams choosing a variant-calling pipeline. nf-core/sarek is designed to be scalable, but actual runtime and resource demands depend heavily on your dataset size, variant caller choice, and compute environment. For example, processing a 30x whole genome sequencing dataset with Strelka2 on a well-configured HPC cluster might take anywhere from 12 to 20 hours, whereas whole exome sequencing runs can complete in as little as 3 to 5 hours. Using more computationally intensive callers, such as Mutect2, or adding extra filtering steps can extend runtimes significantly.
The pipeline’s Nextflow backbone allows it to parallelize tasks efficiently, which is particularly beneficial when running on cloud infrastructure or multi-node HPC setups. Scaling across multiple CPUs or nodes can dramatically reduce wall-clock time, though this comes with increased compute costs on pay-as-you-go cloud platforms. Disk I/O is another factor: alignment and variant calling generate large intermediate files, so ensuring high-throughput storage can prevent bottlenecks.
For labs handling hundreds of samples, batch processing is where nf-core/sarek truly shines, allowing reproducible execution of the same configuration across datasets with minimal manual intervention. However, for smaller labs with limited compute resources, optimizing parameters and selectively running only the necessary steps can help balance turnaround time with resource constraints.
Common Challenges with nf-core/sarek
Even experienced teams can encounter challenges when working with nf-core/sarek. One of the first is the setup overhead: configuring the pipeline to work seamlessly within your specific computational environment, whether it is an HPC cluster, cloud platform, or hybrid system, can require significant time and expertise.
Another common hurdle is the complexity of choosing the right variant caller and setting appropriate filtering strategies. With multiple tools available, each with its own strengths and limitations, deciding on the optimal approach for your study can be daunting.
Finally, in clinical contexts, there is the substantial burden of validation. Since nf-core/sarek is open source and does not come with pre-packaged regulatory certifications, each institution must perform its own verification to meet compliance requirements such as ISO or CLIA. These steps are critical for ensuring accuracy and trustworthiness, but can slow adoption without the right support.
This is where Bionl bridges the gap. We provide pre-integrated nf-core pipelines in a no-code environment, removing infrastructure setup requirements and allowing parameters to be adjusted through a simple interface. This enables teams to focus on analysis and interpretation rather than configuration and troubleshooting.
General Tips and Tricks
Clinical validation:
nf-core/sarekis not validated for clinical use out of the box. Because it is open source, each institution is responsible for validating it in alignment with its specific regulatory requirements. This means that while the pipeline is powerful and flexible, it cannot be deployed in a clinical setting without proper internal validation.Technical expertise required: Running
nf-core/sarekwithout coding knowledge is not straightforward. The pipeline is built on Nextflow and requires some familiarity with command-line operations.Broad compatibility: In terms of compatibility,
nf-core/sareksupports a wide range of sequencing data: whole genome, whole exome, tumor: normal comparisons, and germline datasets across both human and model organisms. This flexibility makes it suitable for a variety of study designs.Platform portability: The pipeline is portable and can run in diverse environments, from local high-performance clusters to cloud platforms such as AWS, Google Cloud, and Azure. It can also be executed through platforms that abstract the backend setup, making deployment far easier.
Final Thoughts
nf-core/sarek offers a rare combination of standardization and flexibility. In research, it is a gold standard for reproducibility. In clinical settings, it is a strong, adaptable base, provided you have the infrastructure for validation.
🔗 At Bionl, we make running nf-core/sarek easier than ever. No coding, no complicated setup; just a streamlined way to explore parameters, scale your workloads, and keep your focus where it matters: on the science.
Related Posts
- Bioinformatics
- nf-core
30–50% Faster in building AI Workflows? Exploring GPT-5 in Bioinformatics
GPT-5 introduces faster responses, smarter routing between model variants, and longer context handling, making it more efficient and reliable than previous versions. In bioinformatics, GPT-5 streamlines workflows by drafting pipelines, configs, and annotations, reducing glue work and acting like a knowledgeable collaborator.
Aug 2025
Abdullah Atia
- Bioinformatics
- No-Code Tools
Bion: Our Multi-Agent Biomedical System
This post introduces Bion, a multi-agent AI system designed to streamline biomedical research by automating data analysis, code generation, and visualization within a no-code notebook interface. Unlike generic AI tools, Bion understands your dataset’s context and uses specialized agents to coordinate complex tasks. It empowers researchers to go from raw data to insights in real time — with or without coding experience. With Bion, science moves faster, smarter, and more intuitively.
Aug 2025